Workflows using Modules
DFT Energy
In terms of practical application, the most popular are calculations of the total electronic energy of a molecule at B3LYP/6-31G(d) level of theory. To carry out such calculations, the following scheme is proposed. This E_B3LYP_631GD workflow assumes simple XYZ input of atomic coordinates.

HF Energy
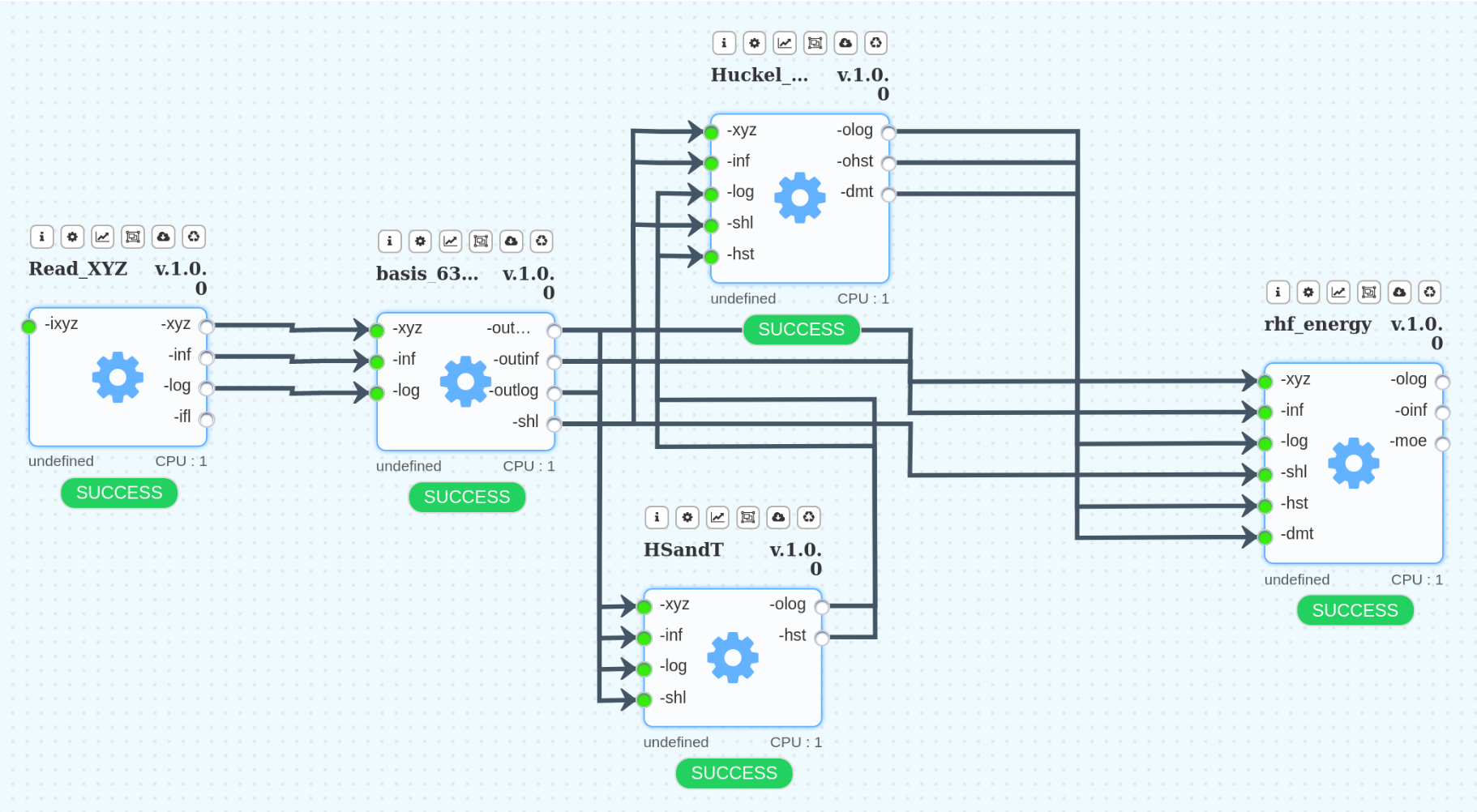
For educational purposes and for comparing the results, the following workflow is useful. This HF_631GD workflow performs computations similar to the above, but at the ab-initio RHF/6-31G(d) level of theory.
B3LYP Energy Basis Effect
Simultaneous energy calculations with B3LYP method but with two different basis sets (6-31G and 6-31G(d)) are available with the E_B3LYP_631G_631GD workflow. Both of them are shown below. A simple XYZ input is needed. The results of simultaneous calculations with two different approach are provided in one output file.

HF vs. B3LYP Computations
Simultaneous energy calculations by RHF and DFT(B3LYP) methods with the 6-31G(d) basis set are available by means of E_HF_B3LYP_631GD workflow. A simple XYZ input is needed. The results of simultaneous calculations with two different approach are provided in one output file.

GAMESS computations
A simple but powerful GAMESS_KMOL workflow allows one to use conventional GAMESS as a built-in module in calculations. The input file can be made both by generating in the graphical mode and uploaded by the user. A simple standard GAMESS input is provided. The user also needs to provide the coordinates of the calculated molecule in XYZ format.

MRSF/TDDFT methods
Computations with the Mixed-Reference Spin-Flip Time-Dependant Density Functional as well as Linear Response Time-Dependant Density Functional methods are available via the built-in GAMESS module. The MRSF_TDDFT workflow below allows one to prepare an input file in a user-friendly way. The input file could by generated in the graphical mode, uploaded by user or the attendant sample input can by used.

Study of effects of inclusion polarization functions in the basis set
Polarization_Function workflow is shown in the figure and it allows to perform simultaneous computations for the same molecule at the DFT (B3LYP) and HF method with two basis sets, one of which, 6-31G, is without polarization functions and the other, 6-31G(d) includes one polarization function for all atoms except for hydrogen atoms. The only geometry of the molecule (.xyz file format) should be provided as an input and four output files are generated as the result.

Gradient and Hessian
Gradient_Hessian_evaluation workflow is shown in the figure and it is designed to perform simultaneous gradient and hessian calculations at RHF/6-31G(d) method. The geometry of the molecule (.xyz file format) is requested as an input and two output files are generated as the result.

Donor-Acceptor interactions
The value of the donor-acceptor interaction can be estimated by comparing the total energy of the donor-acceptor complex and the total energy of its fragments. DAinteraction workflow allows simultaneous B3LYP/6-31G(d) energy calculations of two different fragments of a donar-acceptor complex. Both fragments must have a closed electronic shell. Geometries of both fragments (.xyz file format) should be provided as an input and two output files are generated as the result.

Gradient in different approximations
Gradients workflow allows simultaneous computations of Gradient in different approximations. This performs parallel calculations of the gradient matrix for the same molecule in the DFT(B3LYP) and HF methods with different sets of basis functions(6-31G and 6-31G(d)). The result of the calculations is 4 output files.

Ground and Excited states
Investigation of the excited states of a molecule usually requires a number of formally independent calculations for the same initial geometry of the molecule. Cascade GroundState_2MRSF workflow allows such calculations to be carried out simultaneously. It is assumed that the same initial geometry is used for standard (single point properties or geometry optimization) calculations and for calculations (single point properties or geometry optimization) of the lowest and the second excited states.

Intermediate matrix generation
For both developers’ and educational purposes, it is extremely useful to be able to get the matrices used in standard energy calculations of the electronic structure. The Fock_matrix_Workflow workflow designed to derive the Fock matrix and such its constituents, the matrix of one-electron integrals and the matrix of potential energy. Calculations are available for a wide range of basis sets that can be selected graphically (-i port). The coordinates of the original matrix must be provided in XYZ format (-x port). The results are available in a .dat format file.

SCF procedure
As a base stone of the further development of modular variability, the RHF_SCF_Procedure_Workflow workflow is proposed. It does ab-initio RHF SCF procedure. Calculations are available for a wide range of basis sets that can be selected graphically (-i port). The coordinates of the original matrix must be provided in XYZ format (-x port). The results are available in a .dat format file.
